







The concept of post-extrasystolic potentiation (PESP), which describes the phenomenon of increased contractility of the beat following an extrasystole, has intrigued physiologists and clinicians for more than 120 years. Since its first description in 1885 by Oskar Langendorff,1 PESP has become a widely debated concept, not only for its fundamental basis but also because of the potential diagnostic and therapeutic properties.
Existence of PESP has been demonstrated in isolated papillary muscles,2 perfused isolated hearts3,4 and in vivo models, including humans.5 PESP has formerly been attributed to alterations in preload and/or afterload during the compensatory pause following an extrasystolic beat. However, numerous studies in which preload and/or afterload were controlled,6–9 have demonstrated that PESP is independent of these loading conditions and that its mechanism is the consequence of changes in intracellular calcium handling. A number of reports describe differences of PESP magnitude in patients with heart failure and state that PESP could be used as a marker of myocardial dysfunction.10–13 In addition, recent studies by Sinnecker et al. have shown that the presence of PESP of blood pressure could predict mortality in post-MI patients with sinus rhythm or atrial fibrillation.14,15 These results have revived the possible diagnostic or prognostic role of PESP. In this review, the mechanism of PESP with regard to Ca2+ homeostasis will be discussed in normal and in heart failure individuals and the diagnostic and therapeutic consequences will be explored.
Normal Ca2+ Homeostasis
Excitation–contraction coupling (ECC) is the process by which electrical stimulation results in contraction of cardiac myofilaments, which involves sarcolemmal ion currents and various intracellular pathways.16 Ca2+ has been known to be a major element in both electrical and contractile function of cardiomyocytes. In their resting state, cardiomyocytes have a low cytosolic concentration of Ca2+ ([Ca2+]i) of less than 200 nmol/l.17 When a cardiac cell is depolarised, voltagedependent L-type Ca2+ channels (LTCC) at the sarcolemma open, causing an influx of Ca2+ along its electrochemical gradient into the dyadic cleft. This small inflow of Ca2+ results in release of Ca2+ from the adjacent sarcoplasmic reticulum (SR) through the SR Ca2+ release channels, also known as type 2 ryanodine receptors (RyR2), a process called Ca2+-induced Ca2+-release (CICR). Synchronised opening of RyR2 will generate a global Ca2+ transient,18,19 which increases [Ca2+]i tenfold. Free Ca2+ binds to troponin C, causing a conformational change, which allows the myosin head to bind to actin and move along the actin filament, shortening the cardiomyocyte.
For relaxation to occur, Ca2+ needs to be dissociated from troponin C and be removed from the cytosol. In human cardiac cells, approximately 70 % of cytosolic Ca2+ is sequestered back into the SR by SR-Ca2+ ATPase 2a (SERCA2a) and 30 % is extruded out of the cell by the sarcolemmal Na+-Ca2+ exchanger (NCX), which expels one Ca2+ in exchange for three Na+ ions, creating an inward current.20
Calcium Handling in Heart Failure
Disruption of Ca2+ homeostasis is an important contributor to depressed ventricular function in heart failure. Alterations of Ca2+ uptake, storage and release will result in a reduced Ca2+ transient and consequently a diminished contraction.21–23 Multiple studies suggest that Ca2+ uptake in the SR is diminished due to downregulation or decreased activity of SERCA2a.24–27 A reduced reuptake results not only in decreased SR Ca2+ content but also in higher cytosolic Ca2+ concentration, which inhibits normal relaxation. Therefore, decreased SERCA2a expression contributes to both systolic and diastolic ventricular dysfunction.
On the other hand, expression of NCX appears to be increased in patients with heart failure.28 When functioning in forward mode (Ca2+ efflux and Na+ influx), upregulation of NCX results in increased extrusion of Ca2+ and a decrease of [Ca2+]i. While this may counter the diastolic dysfunction caused by decreased Ca2+ reuptake, there is further reduction in systolic function, due to a decrease in intracellular Ca2+ available for ECC.29
Next, hyperphosphorylation of RyR2 by proteinkinases, such as CaMKII or PKA, is suggested to cause diastolic Ca2+ leakage from the SR.30,31 This Ca2+ leak results in partial depletion of SR Ca2+ stores and contributes further to high diastolic [Ca2+]i. In addition, a diastolic Ca2+ leak may induce Ca2+ release by activating other RyR2, resulting in Ca2+ waves. When Ca2+ is exchanged for three Na+ by the upregulated NCX, a transient inward current (Iti) is generated. This may result in delayed afterdepolarisations (DAD), which could trigger lethal ventricular arrhythmias. Sensitivity of RyR2 for luminal Ca2+ appears to be enhanced; the set point for Ca2+ release is decreased; therefore, RyR2 are activated at lower SR Ca2+ levels in heart failure compared with normal hearts.32 This sensitisation of RyR2 might be an adaption to the decreased Ca2+ concentration in order to maintain normal Ca2+ transients.33 All of these alterations of Ca2+ homeostasis influence both long-term force–frequency relationship (FFR) and short-term force–interval relationship (FIR).
Force–Frequency and Force–Interval Relationship
FFR and FIR describe contractility changes when the stimulation rate is varied. While the FFR describes an altered force of contraction,when heart rate increases or decreases, FIR accounts for change in contractile force by abrupt variations in stimulation pattern, i.e. by introducing extrasystolic beats.
Force–Frequency Relationship
In most mammalian species, including humans, a positive FFR exists in which contractility is enhanced when stimulation frequency is increased.34 This positive staircase phenomenon or ‘treppe’ was first described by Bowditch in 187135 and is an important mechanism for increased inotropy during exercise.36,37 The rise of contractile force appears to be related to an increased amplitude of the Ca2+ transient at higher frequencies.38,39 This increase in Ca2+ transient is the product of different mechanisms. First, an increased number of depolarisations leads to more Ca2+ influx per unit of time, which results in increased calcium release and uptake in the SR. Next, when increasing the frequency of stimulation, the influx of Na+ during depolarisation is increased. To maintain a low cytosolic concentration of Na+, NCX will switch to its reverse mode to extrude Na+ in exchange for Ca2+.40 This influx of Ca2+ will further increase SR Ca2+ content. Finally, reuptake of Ca2+ by SERCA2a relative to extrusion of Ca2+ by NCX is increased.41 This increased SERCA2a activity might be caused by phosphorylation of phospholambam, the main regulatory protein of SERCA2a. In a dephosphorylated state, phospholambam decreases SERCA2a-affinity for Ca2+. When phospholambam is phosphorylated, this inhibitory effect is removed and reuptake of Ca2+ is enhanced. In failing myocardium, FFR is blunted or even inversed, resulting in a decrease of contractile force with increasing frequency of stimulation.34,42–44 This negative FFR is attributed to decreased Ca2+ reuptake due to downregulation of SERCA2a and upregulation of NCX in failing hearts.45,46 When the stimulation rate increases, time per cycle for Ca2+ reuptake is reduced, which, in the case of fewer Ca2+ pumps, results in insufficient Ca2+ reuptake.
Force–Interval Relationship
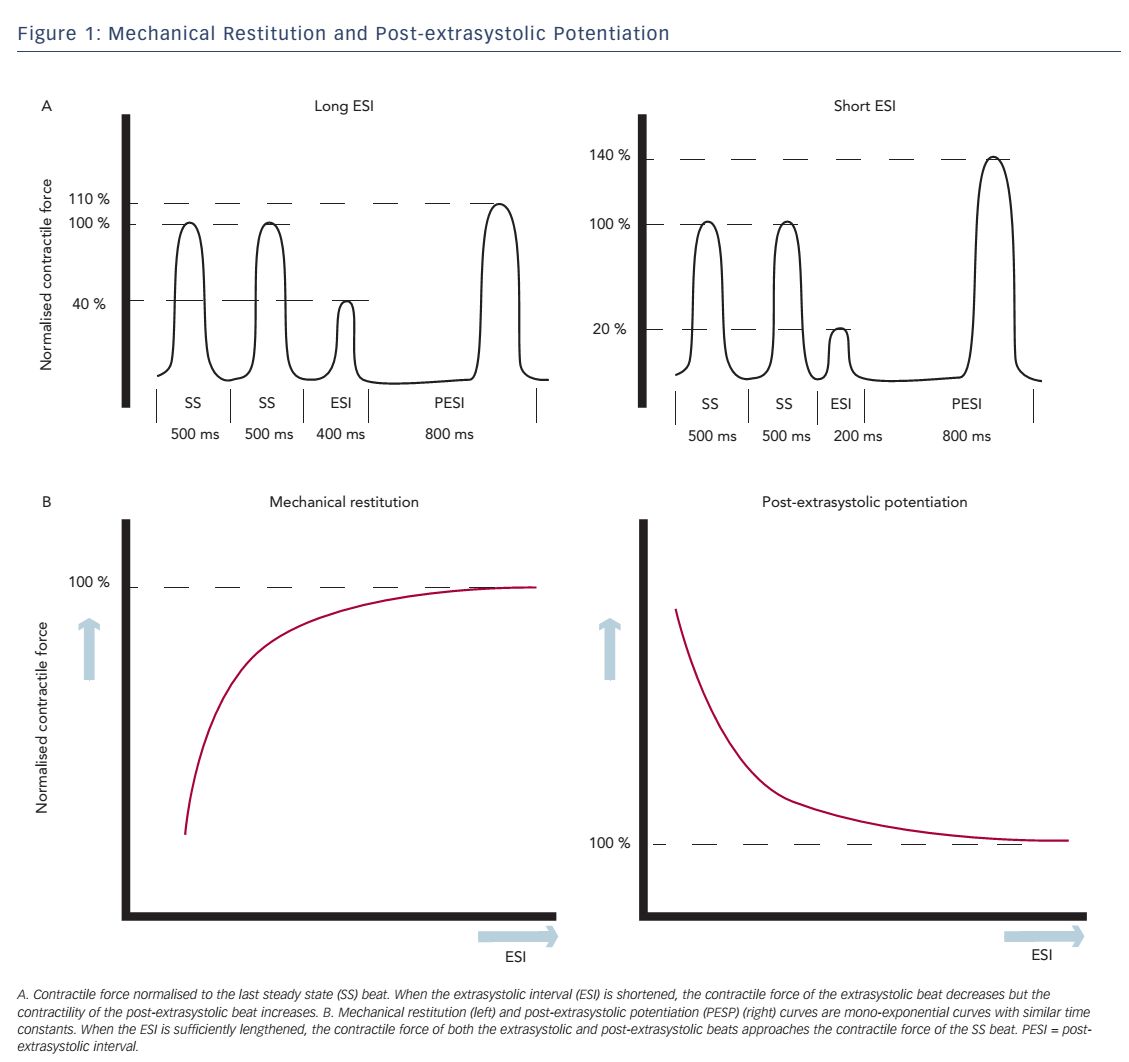
Change in contractile force, when stimulation frequency is interrupted with a premature beat, is described in the short-term FIR. FIR is divided into two concepts: mechanical restitution (MR) and PESP.2 These phenomena are both related to the coupling interval between the regular beat and the premature beat, the extrasystolic interval (ESI), and the interval between the extrasystolic beat and the following postextrasystolic beat, the post-extrasystolic interval (PESI). MR accounts for recovery of contractile strength of the extrasystolic beat when ESI is lengthened. PESP displays the opposite behaviour: with decreasing ESI, there is an increase in contractility of the post-extrasystolic beat. In other words, the earlier the extrasystolic beat occurs, the weaker the extrasystolic beat and the stronger the post-extrasystolic beat (see Figure 1A).
Wier and Yue performed pacing experiments with isolated papillary muscles from ferret hearts to demonstrate the concepts of MR and PESP and the relationship with the ESI.2 After a steady-state pacing series, an extrasystolic stimulus was introduced with varying ESI. The PESI was held constant. As expected, contractility of the extrasystolic beat increased and contractility of the post-extrasystolic beat decreased, when ESI was prolonged. When the contractile strength of the extrasystolic beat and the post-extrasystolic beat was plotted as a function of ESI, monoexponential functions were found with similar time constants (see Figure 1B), which indicates a common underlying mechanism for both phenomena. Nowadays the mechanism of these effects is attributed to changes in Ca2+ handling.
Mechanism of Mechanical Restitution and Post-extrasystolic Potentiation
A fundamental concept for the mechanism of MR and PESP is a time-consuming recovery period of Ca2+ release. The mechanism was formerly explained by a model of different Ca2+ compartments within the SR, in which diffusion of Ca2+ from uptake compartment to release compartment was time dependent.2,3 However, this model lacks experimental evidence, since no anatomical compartment structures have been found in the SR, and transfer by diffusion of Ca2+ within the SR would occur rapidly.47,48
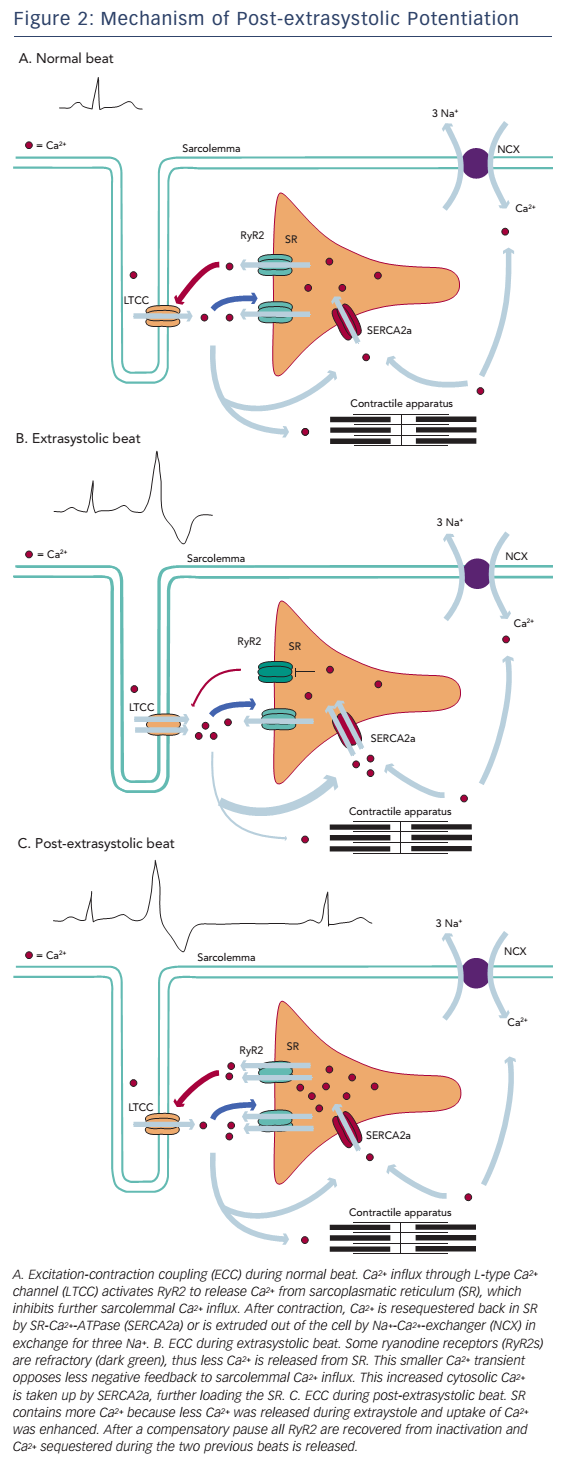
Refractoriness of Ca2+-release channels has been postulated as an alternative explanation for the process of MR and PESP (see Figure 2). In a study by Fabiato,49 recovery from inactivation of ryanodine receptors had a time constant, which was similar to the kinetics of MR and PESP curves by Wier and Yue. According to this model, when a premature beat occurs, most of the ryanodine receptors are refractory to activation, causing a diminished Ca2+ transient and thus a less forceful contraction. After the premature beat, SR Ca2+ load is increased in a number of ways. First, while less Ca2+ is released, Ca2+ loading of the SR continues. Next, low Ca2+ transient during the premature beat opposes less negative feedback to sarcolemmal Ca2+ influx and this extra Ca2+ further increases SR Ca2+ content.
During the compensatory pause there is full MR, thus all release channels have recovered from inactivation. At the post-extrasystolic beat, all the Ca2+ sequestered during the previous two beats will be released, resulting in increased force of the post-extrasystolic beat.
Post-extrasystolic Potentiation in Heart Failure
A number of studies have found differences in PESP of the first derivative of left ventricular pressure (LV dP/dtmax) between heart failure patients and controls. In 1971, Beck et al. observed that patients with obstructed or failing ventricles had an increased potentiation of the post-extrasystolic contraction compared with controls.10 This paradoxical observation was confirmed in other studies.11–13 However, only the study by Seed et al. controlled all coupling intervals (ESI, PESI), which, as we have seen, influence the extent of potentiation.13 Despite the methodological flaws of these clinical studies, a small number of experimental and modelling studies are supportive of this observation.
The increase in PESP is attributed to abnormal Ca2+ homeostasis in heart failure. First, abnormal Ca2+ sequestration could result in a higher PESP. In the study of Seed et al. an inverse linear relationship was seen between PESP and the so-called ‘recirculation fraction’, the ratio of contractility of the second post-extrasystolic beat compared with the first post-extrasystolic beat. The recirculation fraction has been suggested to account for the fraction of released Ca2+ sequestered back into the SR. Patients with heart failure appeared to have a lower recirculation fraction, which might be related to decreased Ca2+ reuptake seen in these patients.
Studies by Hoit et al.50,51 confirmed these results. They evaluated MR and PESP in mice with overexpression of phospholambam, in which Ca2+ reuptake was diminished and recirculation fraction was decreased. They found slower MR and increased PESP in these mice compared with isogenic controls, which indicates a role for SR Ca2+ reuptake in these FIRs. The authors hypothesised that lower SR Ca2+ content slows down recovery of RyR2. When more ryanodine receptors are refractory during the premature beat, even more Ca2+ remains in the SR. During the post-extrasystolic beat the build-up of Ca2+ is released, which results in a higher PESP.
Another explanation for the augmented potentiation in heart failure might be found in an increased sensitivity of RyR2 for Ca2+. In case of more sensitive ryanodine receptors, a larger fraction of the SR content will be released during the post-extrasystolic beat, resulting in an even higher relative PESP in the failing heart.
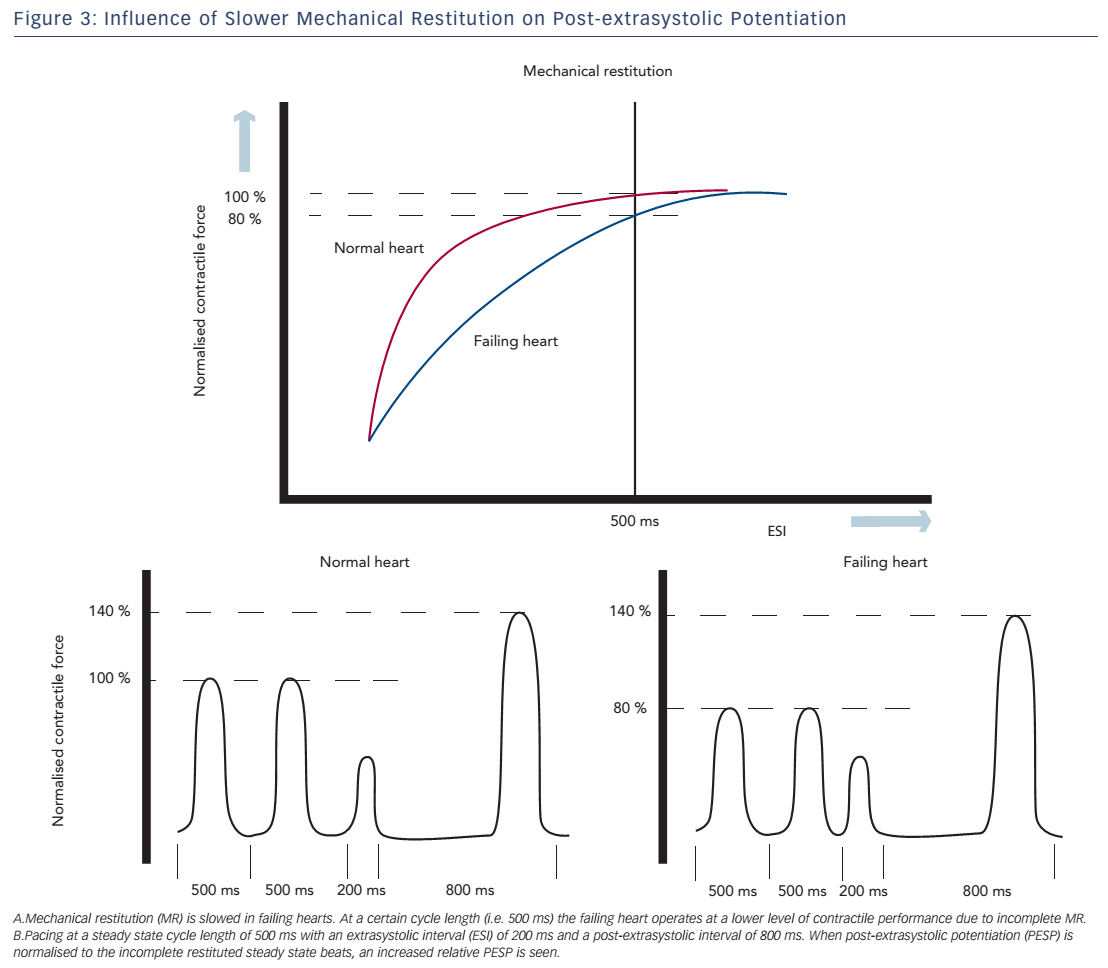
Both of these hypotheses are supported by a study of Rice et al., in which the experiments of Wier and Yue were simulated using a computational model to address different aspects of the short-term FIR.52 The model computed the effects on MR and PESP when certain parameters of ECC were changed. In this model, PESP increased, when the releasable fraction (i.e. the fraction of total Ca2+ in the SR that is released) was increased. This is in accordance with increased sensitivity of RyR2 as an explanation for higher PESP. Furthermore, a decrease in recirculation fraction (the fraction of Ca2+ sequestered back in SR) was also associated with a higher PESP. While this may sound counterintuitive, a lower recirculation fraction results in higher beat-to-beat variability of SR Ca2+ load, which is essential for MR and PESP. In contrast, a theoretical maximal recirculation fraction (all released Ca2+ is recirculated back in the SR) will cause the same SR load of every beat and which makes potentiation impossible to occur. Finally, overall slower recovery of RyR2 may attribute to the increase in PESP. Prabhu et al. investigated alterations of both MR and PESP in dogs with tachycardia-induced heart failure.48,53 They found slower MR kinetics, which they attributed to slower recovery of RyR2. Thus, at faster heart rates, the failing heart does not operate at optimal performance, because most of the ryanodine receptors are refractory, resulting in incomplete MR. When the cycle length is increased, there is full restitution and contractility will return to normal. This observation is consistent with the negative FFR seen in heart failure.
These altered MR kinetics will consequently have implications on PESP. When the failing heart is stimulated at a steady state cycle length below that at which full restitution is achieved, the contractile response of these steady state beats will be suboptimal. During the compensatory pause, the heart is fully restituted and a normal PESP is seen. However, if the magnitude of the post-extrasystolic beat is normalised to the (suboptimal) steady state beats, a higher relative PESP will be found in the failing heart compared with controls (see Figure 3).
Post-extrasystolic Potentiation as a Diagnostic Instrument or Therapeutic Intervention
Diagnostics
PESP has been studied formerly as a diagnostic instrument to differentiate viable from non-viable myocardium during revascularisation procedures.54–56 However, this diagnostic approach has not demonstrated consistent results and has largely been replaced by more accurate techniques, such as nuclear imaging or MRI.
Recently, PESP has gained new attention as a possible prognostic marker in MI patients.14 Sinnecker et al. measured PESP of arterial blood pressure using a non-invasive photoplethysmographic device in 941 patients who survived the acute phase of MI and correlated the presence of PESP to all-cause 5-year mortality. PESP was defined as an increase in post-extrasystolic pulse pressure of 3 % or more compared with the mean of the subsequent beats. The authors found a significant higher mortality risk in patients, in whom PESP was present compared with patients, in whom PESP was absent. PESP remained a significant risk predictor after adjusting for left ventricular ejection fraction (LVEF), the amount of ventricular premature beats and GRACE (Global Registry of Acute Coronary Events) score. Addition of PESP to LVEF as risk predictor increased the area under the ROC curve from 0.61 to 0.75 (P<0.001), indicating that the combination of PESP and LVEF could better stratify patients with high- or lowmortality risk.
The mechanism on how PESP is correlated to a worse prognosis was not made clear. The endpoint all-cause mortality was not further stratified in death of mechanical or arrhythmic origin. As one could assume, changes in PESP displays alterations in Ca2+ handling, which could, in addition to myocardial dysfunction, also lead to early depolarisations and DAD. Thus, altered PESP might indicate an early stage of heart failure, but may also be a marker for increased risk of lethal ventricular arrhythmias.
However, some methodological remarks have to be made. First, the rise in blood pressure of the post-extrasystolic beat was compared with the subsequent beats. However, PESP usually decays in a number of beats, therefore, for correct analysis of the percentage of PESP, using the beats preceding the premature ventricular contractions (PVCs) would have been more accurate. Second, in this study PESP was defined as difference in blood pressure, measured with a non-invasive device at the finger, while most studies used invasive measurements of contractility, such as the first derivative of LV pressure (dP/dt). When measuring PESP more distally, vascular influences might alter the blood pressure measurements of PESP. In other words, the phenomenon measured in this study might not be comparable to PESP seen during earlier invasive experiments.
More importantly, as seen in other studies of PESP in heart failure, the intervals were not held constant. Therefore, when basic rhythm, extrasystolic intervals or PESI differ, the magnitude of potentiation will change.57,58 Therefore, from a physiological point of view, no strong conclusions can be made on the relationship between high PESP and heart failure, when the intervals are not controlled. Nonetheless, since PESP appears to be a strong predictor of mortality, it could still have prognostic value, even if the underlying mechanism is not completely understood. Further validation of the use of PESP as a prognostic marker will be needed before it could be implemented as a clinical tool.
Therapeutics
The therapeutic use of PESP has been extensively investigated in the 1960s and 1970s, but has long been abandoned due to conflicting evidence of its effectiveness.59–62 Inducing PESP, and therefore increasing the force of contraction, might be beneficial in heart failure patients. By using coupled or paired pacing, in which a premature beat is introduced after every other intrinsic or paced beat, respectively, the effect of PESP is extended, which improves contractility and cardiac output. This technique has been studied in patients with cardiogenic shock, in which cardiac function significantly improved. However, reports of increased myocardial oxygen consumption and risk of arrhythmias have reduced the interest in this mode of pacing.63
More recently, a number of studies have reevaluated the safety and efficacy of coupled pacing. In 2008, a study by Lieberman et al.64 studied dual chamber coupled pacing (DCCP) in 16 heart failure patients. DCCP increased LV dP/dtmax and arterial pulse pressure; however, other haemodynamic parameters, such as mean arterial pressure, cardiac output and mixed venous O2 saturation did not differ.
In the same year, Freudenberg investigated the use of atrioventricular coupled pacing in 10 heart failure patients and concluded that this mode of pacing is safe and well tolerated.65 In this study, coupled pacing was applied over 15–20 minutes. A significant increase in EF and stroke volume and a reduced end systolic volume were seen, accompanied by a decrease in cardiac output due to a decreased pulse rate.
Two studies evaluated the use of coupled pacing in addition to cardiac resynchronisation therapy in heart failure patients with mechanical dyssynchrony.66,67 A further increase in contractility and EF was seen along with a decrease in pulse rate without disrupting the synchronisation properties of CRT. While Stegeman et al. concluded that this drop in pulse rate reduced the haemodynamic benefit of paired pacing, Brémont et al. suggested that the heart rate reduction could have an additional beneficial effect because of increased time for ventricular filling and reduced myocardial work.66,67
These recent trials indicate that there still might be a role for coupled pacing in heart failure patients; however, the long-term effects remain unknown. Furthermore, because of introduction of stimuli during the vulnerable period of repolarisation, the risk of inducing ventricular arrhythmias remains present. Another device-based therapy, cardiac contractility modulation (CCM), is increasingly being investigated and has already shown to be a safe and effective alternative to coupled pacing. CCM uses high-intensity, non-excitatory electrical signals applied during the absolute refractory period, which, in contrast to PESP, do not result in an action potential nor contraction.68–70 Studies in isolated papillary muscles,71 isolated hearts,72 in vivo animal models73 and patients74 have shown a positive effect on cardiac contractility. This treatment might be a good option for patients with advancing heart failure despite optimal medical treatment who are not a candidate for CRT. The exact mechanism of action is unknown, but the effect has been attributed to an increase in Ca2+ transient by a number of possible mechanisms, including an increase in phosphorylation of phospholambam, increased SERCA2a expression75 and normalisation of NCX activity,76 but also an increased influx of Ca2+ through the L-type calcium channels.69 A number of randomised clinical trials77–79 have been executed to investigate the safety and efficacy of CCM in heart failure patients and showed an improved exercise tolerance and quality of life, without increased myocardial oxygen consumption or arrhythmia risk. However, no difference in mortality nor morbidity have yet been found, thus long-term consequences need to be further elucidated before these techniques can be implemented in clinical practice.
Conclusion
Since it was last reviewed extensively in 1993 by Cooper, much has been discovered about the phenomenon of PESP.80 The fundamental physiology of an altered calcium homeostasis has been further elucidated; the SR compartment model of Wier and Yue has been replaced by the central role of the ryanodine receptor and its refractory period in the mechanism of PESP.
The diagnostic and therapeutic properties of PESP have recently been rediscovered. However, the relationship of PESP and heart failure remains a complex interplay of both FIR and FFR. Therefore, experimental studies of control versus failing hearts, in which all intervals are controlled, will be needed to confirm the assumption of an augmented PESP in failing hearts. In addition, the prognostic value of PESP of blood pressure needs to be further evaluated and validated, since this non-invasive test might be of great value for selecting therapeutic interventions, e.g. ICD therapy, in certain patient groups. The Comparative Effectiveness Research to Assess the Use of Primary Prophylactic Implantable Cardioverter Defibrillators in Europe (EU-CERT-ICD) trial, which investigates new parameters for identification of high arrhythmia risk in ICD patients, is currently ongoing and will incorporate the use of PESP for stratification of mortality and ICD shock risk. Finally, the results of trials on the therapeutic use of coupled pacing in conjunction with CRT therapy have again attracted attention. However, CCM might take its place as a new device-based therapy for heart failure, because of a better safety profile. Further evaluation of the long-term effects of this new therapeutic option will be needed to confirm these promising results.