







Hypertrophic cardiomyopathy (HCM), the most common genetic cardiomyopathy, is present in one in 500 of the general population and is caused by over 1,400 mutations in at least 11 genes encoding the cardiac sarcomere.1–4 Although the majority of patients with HCM remain asymptomatic with near-normal longevity, a small, but important, subset of patients are at increased risk for a wide range of clinical outcomes including development of advanced heart failure symptoms, atrial and ventricular arrhythmias, thromboembolic events, and even sudden death.5–8 HCM is characterised by a heterogeneous phenotypic expression with diverse range of extent and pattern of hypertrophy (massive to minimal hypertrophy, that can occur at any location from the apex to the base),4,9 outflow obstruction (resting, provocable or nonobstructive),10 and left ventricular (LV) systolic function (hyperdynamic to systolic dyfunction).1 Cardiovascular magnetic resonance (CMR), a highresolution 3D tomographic imaging technique that provides sharp contrast between the blood pool and myocardium, has emerged as an imaging technique that is particularly well suited to characterise the diverse morphological expression of this disease (see Figure 1), and is the imaging modality of choice when the diagnosis or morphological characteristic of HCM remains in doubt following echocardiography.1,2,11–13 In addition, contrast-enhanced CMR with late-gadolinium enhancement (LGE) has the capability to identify areas of myocardial fibrosis/scarring with novel data demonstrating that the extent of LGE by CMR may play an important role in risk stratification of patients with HCM.14–18 Thereby, it is timely to discuss the specific areas that CMR contributes in the clinical evaluation and risk assessment of patients with HCM.
Diagnosis
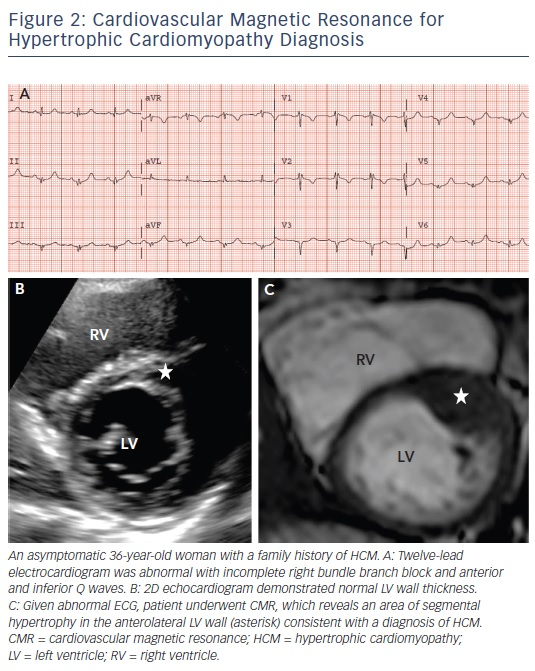
A diagnosis of HCM is made when unexplained LV hypertrophy (range 13–60 mm; mean 22 mm) occurs in the absence of another disease capable of producing a similar magnitude of hypertrophy.5,6 Therefore, the clinical diagnosis is highly dependent on accurate non-invasive quantification of the LV wall thickness. Traditionally, 2D echocardiography has been the primary imaging modality used in evaluation; however, the echocardiographic examination may provide measurements that appear to fall within the non-diagnostic range (i.e. normal or borderline increase).9 By virtue of its high spatial resolution, CMR allows a more precise assessment of LV wall thickness and areas of hypertrophy. In fact, CMR has identified focal and segmental areas of hypertrophy within the LV that is not reliably identified by 2D echocardiogram, particularly in the anterolateral free wall, apex or posterior septum (see Figure 2).9,19 This is an important consideration as 20 % of patients with HCM have focal areas of hypertrophy, confined to one or two LV segments.4 For these reasons, when a clinical diagnosis of HCM is suspected due to clinical symptoms, electrocardiographic abnormalities or family history, and echocardiography is normal/non-diagnostic, additional testing with CMR should be performed.5
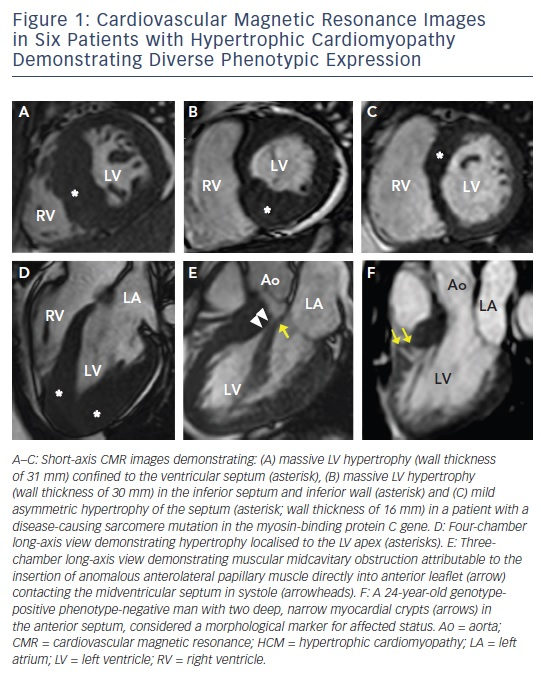
Areas of LV hypertrophy may similarly be underestimated by echocardiography, with more accurate measurements made by CMR. This has important management implications as massive hypertrophy (wall thickness ≥30 mm) is an independent risk factor for sudden death in HCM, and in some patients may only be recognised by CMR.2
Similarly, an overestimation of LV wall thickness may also occur with echocardiography. For example, when the crista supraventricularis, a right ventricular muscle structure, is situated adjacent to the ventricular septum; this structure may be inappropriately included in the septal measurements by echocardiography, an overestimation of wall thickness that can be avoided using CMR.13
Assessment of Family Members with Hypertrophic Cardiomyopathy
Screening of all first-degree relatives of patients with HCM is indicated to identify those individuals with potentially unrecognised disease.5,6 Screening should begin at the onset of adolescence, with repeat imaging performed annually (every 12–18 months) throughout adolescence, and then every 5 years until the fourth decade of life, as delayed-onset hypertrophy can also occur later in adulthood. While echocardiography has traditionally been the mainstay test used in screening, the realisation that CMR provides a more precise delineation of LV hypertrophy has led to the increased use as part of the screening evaluations.20,21 This not only allows for more accurate diagnosis, but also a benchmark for future studies to better define the potential progression of LV hypertrophy.
The availability of genetic testing in clinical practice has resulted in the identification of family members with HCM who carry a diseasecausing sarcomere mutation (and therefore are at risk of developing phenotypic HCM), but without LV hypertrophy (i.e. genotype positive– phenotype negative [G+P−] patients).20–23 This led to the observation with echocardiography that abnormalities of myocardial function are present in G+P− patients, and the emerging principle that even in the absence of increased LV wall thickness these hearts may be abnormal.21–23 CMR has added to these insights by demonstrating that a number of additional morphological abnormalities may be present including myocardial crypts (see Figure 1F), elongated mitral valve leaflets, expanded extracellular space (with T1 mapping) and LGE.24–27 When genetic testing is negative or ambiguous (as in 60 % of patients), or when not pursued due to financial or personal preference, CMR can identify these abnormalities in the absence of LV hypertrophy, raising suspicion for genotype-positive status among family members.2,21 This should prompt continued close surveillance with serial CMR for development of LV hypertrophy and conversion to clinical disease.
Differentiation of Other Aetiologies of Left Ventricular Hypertrophy
Athlete’s Heart
LV hypertrophy associated with systemic training (i.e. athlete’s heart) may be difficult to differentiate from HCM.28,29 The differentiation between athlete’s heart and HCM is critical as HCM is an important cause of sudden death in athletes, responsible for 6–36 % of events.30–32 A variety of different morphological features on CMR may help distinguish HCM from athlete’s heart. Additionally, CMR can evaluate for other structural abnormalities that are also frequently implicated in sudden death of athletes including arrhythmogenic right ventricular cardiomyopathy and myocarditis.30–32 Thereby, a normal CMR provides a further level of reassurance.
CMR can help differentiate athlete’s heart from HCM by identification of focal pattern of hypertrophy, a finding supportive of a diagnosis of HCM. In addition, forced deconditioning of an athlete may serve as a useful strategy to resolve diagnosis, with CMR well suited to compare maximum LV wall thickness measurements before and after a period of systemic deconditioning. In this regard, a patient whose wall thickness regresses by more than 2 mm supports a diagnosis of athlete’s heart, while hypertrophy that remains present despite deconditioning supports a diagnosis of HCM.33
Contrast-enhanced CMR with LGE, provides the opportunity to aid in the differentiation between HCM and athlete’s heart given the ability to non-invasively provide tissue characterisation by means of identifying focal areas of replacement fibrosis and expanded extracellular space. While LGE is present in about half of individuals with HCM, LV remodelling associated with athlete’s heart should not result in focal areas of myocardial scarring/fibrosis, especially in younger individuals.33 Therefore, in an athlete suspected to have HCM, the presence of LGE on contrast-enhanced CMR favours a diagnosis of HCM. In contrast, the absence of LGE cannot be used to reliably exclude the possibility of HCM as this is found in half of patients with a clinical diagnosis of HCM.13
Hypertensive Cardiomyopathy
The differentiation of LV hypertrophy due to systemic hypertension from HCM has historically been challenging. CMR can help in differentiation by examining the pattern of hypertrophy, with longstanding systemic hypertension resulting in more concentric hypertrophy (near-identical hypertrophy in septum and lateral wall), while LV wall thickening in HCM is more commonly asymmetric.11–13 This asymmetric pattern favours a diagnosis of HCM over hypertension; however, it should be noted that in some patients with HCM the pattern of hypertrophy may also be symmetrical.11–13 Additionally, presence of LV outflow obstruction due to typical systolic anterior motion of the mitral valve will help sway a diagnosis towards HCM, as this finding is present in over two-thirds of patients with HCM and rarely seen in hypertensive cardiomyopathy.11–13 CMR can also be helpful in the detection of changes in serial measurements of LV wall thickness after aggressive treatment with antihypertensives, in which a regression of hypertrophy would favour a diagnosis of hypertensive cardiomyopathy.
Infiltrative Cardiomyopathy
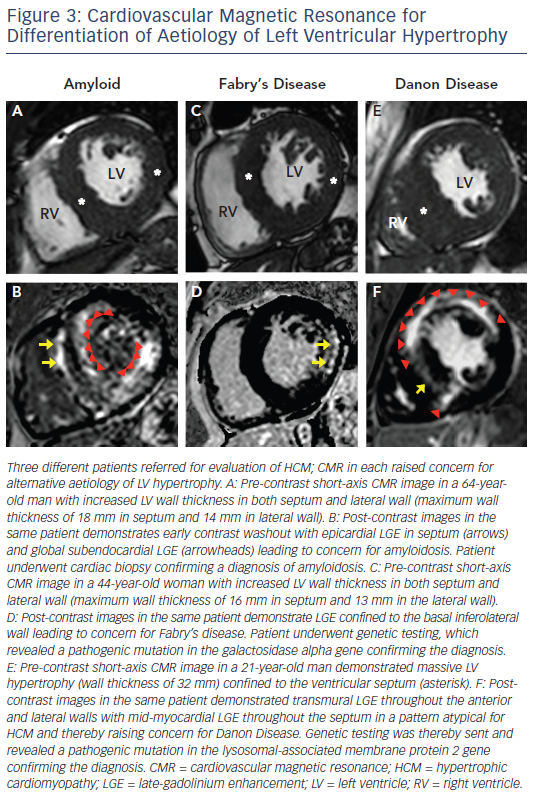
Infiltrative cardiomyopathies, including amyloidosis or glycogen/ lysosmal storage diseases (such as Fabry’s or Danon disease) can mimic clinical HCM as they can produce increased wall thickness as part of their phenotypic expression (see Figure 3).34–36 Although these diseases may have non-cardiac signs and symptoms, disease expression can also be confined only to the heart. The accurate differentiation of these ‘phenocopies’ is critical as treatment strategies and prognosis differs compared with HCM. In amyloidosis, CMR identification of increased LV wall thickness in both the lateral wall as well as the septum combined with global subendocardial LGE is suggestive of cardiac amyloidosis and not typical in HCM.34 Suspicion of Fabry’s disease, an X-linked storage disease in which mutations in the alpha-galactosidase A gene leads to cellular accumulation of glycosphingolipids in multiple organs including the heart, and potentially treatable with enzyme replacement therapy, can be raised by increased LV wall thickness in both the lateral wall and septum with LGE confined to the basal inferolateral wall.35 Danon disease, which is due to mutations in genes that encode the lysosomal-associated membrane protein 2, leads to accumulation of intracellular vacuoles and is a profound and accelerated disease process with rapid clinical deterioration leading commonly to advanced heart failure and sudden death at a young age (commonly <25 years old).36 CMR can be suggestive of the diagnosis in the setting of massive LV hypertrophy with extensive diffuse and often transmural LGE.37 T1 mapping, a novel CMR sequences, has potential to help in the further differentiation of HCM from these infiltrative cardiomyopathies.38,39 Although CMR findings may be suggestive of a phenocopy in a patient undergoing evaluation for HCM, CMR findings in themselves are not diagnostic and must be considered within the clinical contest of an individual patient. Therefore. confirmation with either laboratory testing, molecular genetic analysis or biopsy (either cardiac or another affected tissue) is often ultimately required to make a definitive diagnosis.5,6
Phenotype Characterisation of HCM
Left Ventricular Apical Aneurysms
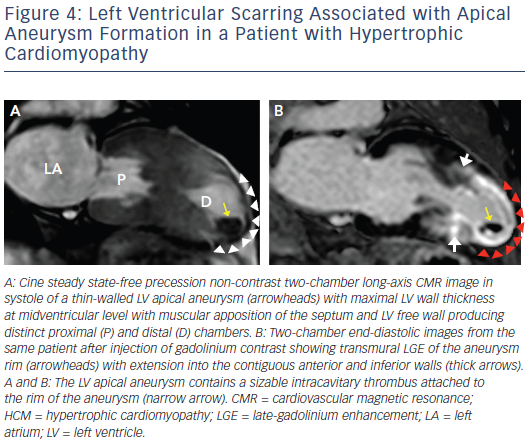
Increasing penetration of CMR into routine cardiovascular practice has resulted in more frequent identification of a subset of patients with an unusual phenotype of HCM with thin-walled, scarred LV apical aneurysms (see Figure 4). This important group of patients had been underdiagnosed prior to the application of CMR to HCM, largely based on small- to moderate-sized aneurysms not reliably identified by echocardiography.40 Contrast-enhanced CMR has demonstrated that the aneurysm rim in these patients is composed predominantly of fibrosis that extends from the aneurysm rim into the septum and free wall and serves as nidus for ventricular tachycardia. These changes may place in patients at increased risk of arrhythmic sudden death and thromboembolic stroke (secondary to LV thrombus formation in the aneurysmal cavity).40 Thereby, the identification of LV apical aneurysms may raise important management implications with consideration for implantable cardioverter defibrillator (ICD) therapy as well as systemic anticoagulation for stroke prevention.1,5
Outflow Obstruction
Mechanical impedance to LV outflow due to systolic anterior motion of the mitral valve is perhaps the most important cause of limiting heart failure symptoms in HCM.10 The identification of LV outflow obstruction in the setting of drug-refractory severe symptoms is critical as it alters management strategies towards invasive septal reduction therapy with either surgical myectomy or alcohol septal ablation.5,6 CMR allows for precise evaluation of the left ventricular outflow tract (LVOT) and anomalies contributing to outflow obstruction, including anomalous insertion of the anterior papillary muscle directed into the mitral leaflet (see Figure 1e), elongated mitral valve leaflet lengths and muscle bundles that extend from the apex and attach into the basal anterior septum.41,42 The identification of these features may be missed by echocardiography yet are critical as they potentially alter the septal reduction strategy in favour of surgical myectomy, as alcohol septal ablation is unable to address these additional abnormalities.43,44
Risk Stratification
Sudden Death
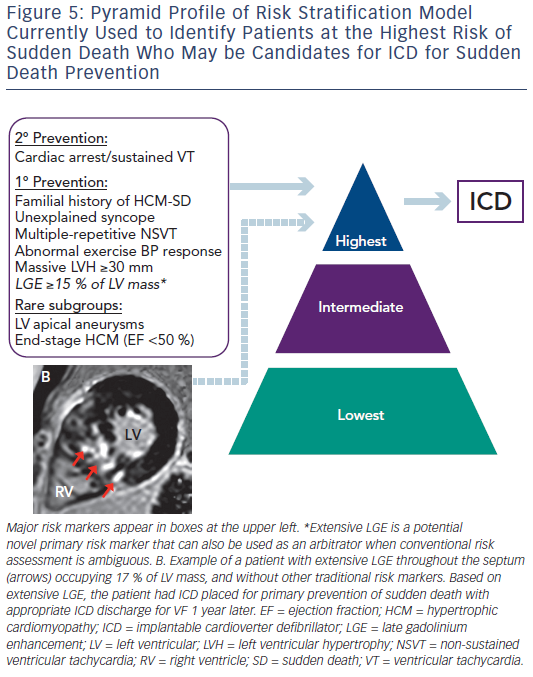
Since the initial descriptions of HCM, sudden death has been a highly visible and devastating disease consequence. Fortunately, sudden death is confined to a small subset of patients with HCM within the broad disease spectrum.7,8 Sudden death events occur unpredictable, often without warning signs or symptoms and is most common in young people through mid-life.1 The application of ICD for primary prevention of sudden death in HCM has created the opportunity to prevent these catastrophic events.45 This has placed increased importance on risk stratification to help identify individuals who may benefit from device therapy for primary prevention. The current American College of Cardiology (ACC) and American Heart Association (AHA)-based HCM risk stratification algorithm has relied on five major risk markers (see Figure 5) and has been highly effective in identifying many patients with HCM who will benefit from ICD therapy.5 While this has been instrumental in decreasing rates of sudden death and HCM-related mortality to 0.5 %/ year, some patients without conventional risk markers nevertheless remain at risk of sudden death.7,8 These limitations have led to an interest in additional strategies to improve the current risk model. In this regard, attention has focused on contrast-enhanced CMR with LGE to non-invasively identify myocardial fibrosis, the potential arrhythmogenic substrate in HCM.14–18 Early studies demonstrated that patients with HCM and evidence of LGE on CMR have increased rates of non-sustained ventricular tachycardia on ambulatory Holter monitoring compared with patients without LGE, raising the concept that LGE represents a substrate for generation of malignant ventricular arrhythmias.14
This notion led to several outcome studies, each with relatively small patient cohorts, evaluating the presence of LGE on CMR and demonstrating that patients with HCM with LGE were at increased risk of cardiovascular mortality.16–18 However, LGE is fairly common in patients with HCM, with a prevalence of >50 %, and thereby the use of presence of LGE alone as a sudden death risk marker would lead to over-implantation of ICD for primary prevention.2
Conversely, a large multicentre study with almost 1300 patients with HCM demonstrated that LGE extent is capable of identifying patients at increased sudden death risk and deserving of consideration of ICD placement.15 Extensive LGE, occupying ≥15 % of LV mass, is equivalent to a twofold sudden death risk as compared with no LGE. This increased sudden death risk is present even among patients without other established risk markers and who would otherwise be considered at low risk. Furthermore, when data from this study was pooled with data from a study by Ismail et al.,46 the only other study to report adjusted hazard ratio for the extent of LGE in HCM, the amount of LGE remains independently associated with sudden death risk (adjusted hazard ratio 1.4 for every 10 % increase in LGE of LV mass; and adjusted hazard ratio of 1.6 for 15 % LGE).47 Based on these data, it may be reasonable to consider that patients with HCM with extensive LGE (≥15 % LV myocardium) at increased risk, independent of other high-risk features, with implications on management strategies including consideration for primary prevention ICD therapy (see Figure 5).2,15
Extensive LGE also helps resolve decision making regarding ICD in complex situations when sudden death risk remains ambiguous after standard risk stratification, as it can serve as an arbitrator towards ICD placement.15 In contrast, the absence of LGE is associated with lower risk for sudden death and should provide a measure of reassurance.2,15 Therefore, LGE has emerged as a potentially powerful tool to strengthen the ACC/AHA risk stratification model.
Systolic Dysfunction
Extensive LGE can also be predictive of progression to the end-stage phase of HCM, characterised by LV remodelling with ventricular cavity dilation, wall thinning secondary to scarring and systolic dysfunction (ejection fraction <50 %).48 Extensive LGE, comprising ≥15 % of total LV mass, also prospectively identifies patients with preserved systolic function who are at risk of heart failure progression due to systolic dysfunction and may require future heart transplantation.15 This recognition can alter management strategies including consideration for altered medical therapy, prophylactic ICD and timely evaluation for heart transplantation once symptoms develop.48
Future Direction: T1 Mapping
T1 mapping is a novel and promising CMR technique that provides assessment of the total extent of expanded extracellular space, rather than the detection of regional areas of myocardial fibrosis identified by traditional LGE imaging.49 It has been postulated that T1 mapping may emerge as a diagnostic imaging marker in differentiating pathological cardiovascular diseases such as HCM from that of other forms of LV hypertrophy (such as Fabry’s disease39 or amyloidosis38) and that this technique may prove to be superior to LGE for risk stratification in HCM. However, to date, there has been no link between T1 mapping and cardiovascular outcomes within HCM. In addition, conflicting data exist regarding T1 mapping values in G+P– patients, and if this value can indeed differentiate G+P– patients to normal controls.24,50 Thereby, continued investigations applying T1 mapping to HCM is necessary to better to define the role of this technique.
Conclusion
Over the last decade, contrast-enhanced CMR has emerged as a powerful imaging tool uniquely suited for the characterisation of the heterogeneous phenotypes in HCM.9–13 CMR provides relevant diagnostic and prognostic information not identifiable with traditional echocardiography.15–19 CMR impacts a variety of clinical management issues ranging from diagnosis and family screening to procedural planning for septal reduction therapy.20,25,38–41 Newer data demonstrate that extensive LGE, occupying ≥15 % LV myocardium, identifies patients at an increased sudden death risk and these patients may ultimately benefit from ICD placement for primary prevention.2,15 These observations help to justify an expanded role of CMR in the routine assessment of patients with HCM.
Clinical Perspective
- Contrast-enhanced CMR has emerged as a power imaging tool uniquely suited for the characterisation of the heterogeneous phenotypes in HCM.
- CMR helps to diagnose HCM given its abilities to identify areas of hypertrophy that is not well visualised by echocardiography, to provide more accurate wall thickness measurements and to differentiate other aetiologies of LV hypertrophy.
- Contrast-enhanced CMR with LGE has identified patients with extensive LGE, occupying ≥15 % LV myocardium. Based on data from a recent large multicentre study, it may be reasonable to consider that these patients are at increased risk of sudden death, independent of other high-risk features, with implications on management strategies including consideration for primary prevention ICD therapy.
- These observations help to justify an expanded role of CMR in the routine clinical assessment of patients with HCM.